A Comprehensive Survey of Protein Forms
Development of Novel Methods for Proteoform Analysis by Top-Down Mass Spectrometry
An international collaborative research group with members from Ehime University, the National High Magnetic Field Laboratory (NHMFL), the University of California, Los Angeles (UCLA), and the University of Liverpool has developed a novel method to rapidly and efficiently recover intact proteins separated by polyacrylamide gel electrophoresis and decode their sequences by mass spectrometry. The results are published in the Journal of Proteome Research. The paper was selected as the ACS Editors' Choice and featured on the journal’s supplementary cover.
Proteins undergo post-translational modification in the in vivo environment to alter their structure and activity to achieve their physiological functions. Therefore, a detailed understanding of the many different forms of proteins (proteoforms) in vivo is crucial for grasping the elaborate workings of biological systems at the molecular level. However, it is still unclear how many proteoforms are ultimately produced by the approximately 20,000 human genes.
Top-down mass spectrometry is an analytical method for decoding the full-length sequences of proteins in detail. In recent years, the Consortium for Top-Down Proteomics has started to use top-down mass spectrometry to comprehensively analyze human proteoforms (Top-Down Proteomics), and as a result, the existence of disease-specific proteoforms has gradually been revealed. However, the complexity of protein components in living organisms has made it a challenge to analyze proteins at low concentrations using top-down mass spectrometry.
In this study, we developed a novel technique for the detailed fractionation of complex protein components extracted from biological samples by SDS-polyacrylamide gel electrophoresis. Furthermore, by combining the developed technology with top-down mass spectrometry, we successfully established a novel top-down proteomics workflow (PEPPI-MS) for the sensitive analysis of small amounts of proteoforms in biological samples.
In collaboration with Dr. Lissa C. Anderson of NHMFL, we developed a high-resolution PEPPI-MS system utilizing the 21 tesla Fourier transform ion cyclotron resonance mass spectrometer at NHMFL, which makes it possible to detect more than 1,000 proteoforms from a very small protein sample extracted from E. coli. Furthermore, by combining PEPPI-MS with native polyacrylamide gel electrophoresis (gel electrophoresis of proteins in a non-denaturing state), we succeeded in recovering protein complexes of interest from biological samples and analyzing their structures by mass spectrometry.
PEPPI-MS will dramatically increase the number of detectable proteoforms in top-down proteomics, which has the potential of leading to the discovery of novel proteoforms. Moreover, PEPPI-MS makes it possible to obtain information on blood proteoforms from minute amounts of blood samples, which can lead to the identification of proteoforms characteristic of diseases and can aid in molecular diagnosis based on the proteoforms.
Reference URL: https://pubs.acs.org/doi/10.1021/acs.jproteome.0c00303
Bibliographic Information
Journal: Journal of Proteome Research
Title: PEPPI-MS: Polyacrylamide-Gel-Based Prefractionation for Analysis of Intact Proteoforms and Protein Complexes by Mass Spectrometry
Authors: Ayako Takemori, David S. Butcher, Victoria M. Harman, Philip Brownridge, Keisuke Shima, Daisuke Higo, Jun Ishizaki, Hitoshi Hasegawa, Junpei Suzuki, Masakatsu Yamashita, Joseph A. Loo, Rachel R. Ogorzalek Loo, Robert J. Beynon, Lissa C. Anderson*, and Nobuaki Takemori*(*, Corresponding authors)
DOI: 10.1021/acs.jproteome.0c00303
Fundings
- the Scientific Research on Innovative Areas “Chemistry for Multimolecular Crowding Biosystems” (JSPS KAKENHI Grant No. 18H04559)
- the Grant-in-Aid for Scientific Research C (JSPS KAKENHI Grant No. 16K08937)
- the Grant-in-Aid for Scientific Research C (JSPS KAKENHI Grant No. 19K05526)
Media
-
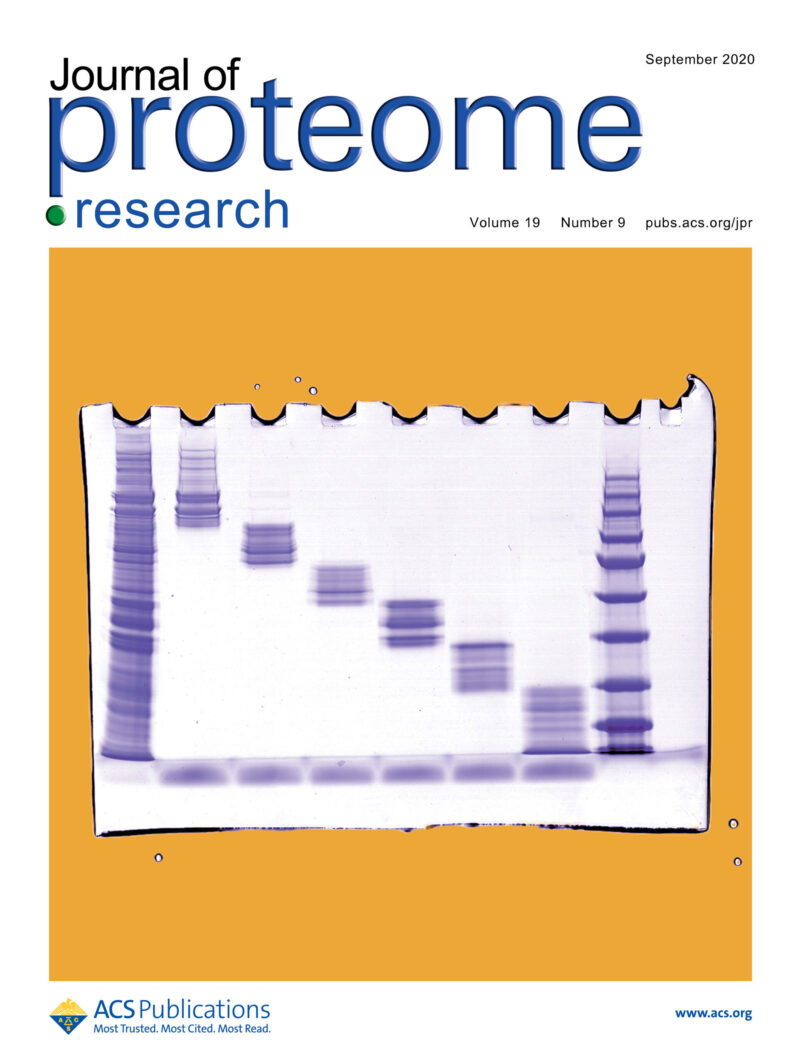
SDS-PAGE image of a PEPPI-MS fraction of the proteome of the Drosophila compound eye (JPR Supplementary Cover).
We established a novel method for rapid and efficient extraction of intact proteins from polyacrylamide gels called PEPPI-MS. This method allows efficient recovery of intact proteins from a wide range of molecular weight regions after high-resolution protein separation by PAGE. The PEPPI-MS workflow would facilitate in-depth top-down proteomics for complex biological samples.
credit : Reprinted with permission from Journal of proteome research. © 2020 American Chemical Society
Usage Restriction : Please get copyright permission
Contact Person
Name : Nobuaki Takemori
Phone : +81-89-960-5499
E-mail : takemori@m.ehime-u.ac.jp
Affiliation : Advanced Research Support Center